Tracking mutation progression on SARS-CoV-2 druggable cavities, epitopes, and binding interfaces
Date:
12/1/2021
Tracking the evolution of the SARS-CoV-2 viral components is
vital for identifying potential efficacy shifts for existing
and novel therapeutics (e.g. the Pfizer mRNA vaccine,
Casirivimab, Indevimab, Bamlanivimab, Remdesivir) as well as
understanding potential causes of increased virulence, e.g.
the destabilising Spike glycoprotein D614G mutation, and
mutations in the UK and South African variants that facilitate
the viral spike activation and viral-host membrane fusion.
Furthermore, viral mutations can affect interactions with
human proteins, thus altering the virus - host interactome.
Such mutations can impact cell signalling and, from the point
of view of drug discovery, may introduce new therapeutic
opportunities via novel virus - host protein interactions or
negate existing ones should mutations diminish established
virus - host protein interactions.
canSARS, utilises canSAR-3D (the unique 3D structural
component of canSAR, that uses artificial intelligence
approaches to identify and predict the 'ligandability' of
proteins with known 3D structure) in conjunction with the
canSARS druggable core networks (Druggable Interactome report), to triage the comprehensive GISAID SARS-CoV-2 mutation
data provided by
CoV-GLUE
with additional literature curation of epitope sites on viral
components (e.g.
Shrock et al., 2020).
In this report we compare and contrast two snapshots of the
mutational landscape of SARS-CoV-2, the first from mid-June
2020 (217,204 protein coding mutations) and the second from
mid-November 2020 (1,197,272 protein coding mutations):
with additional literature curation of epitope sites on viral
components (e.g.
Shrock et al., 2020).
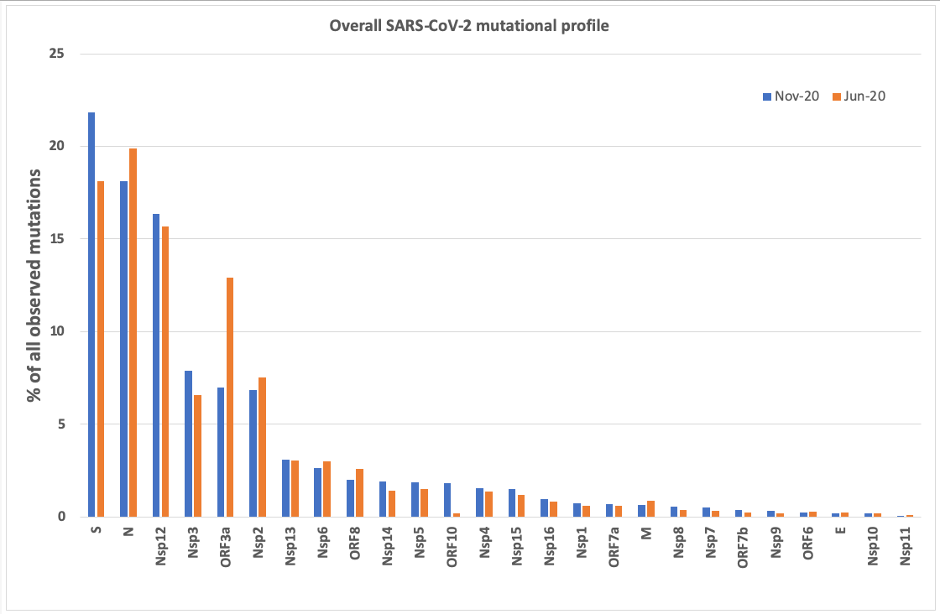 Overall Mutational Profile SARS-CoV-2
Overall Mutational Profile SARS-CoV-2
Despite the significant increase in the reported number of
mutations between the two snapshots, their relative
distribution is comparable, with two notable exceptions: an
3.7% increase in mutations occurring in the Spike glycoprotein
and a 5.9% decrease in mutations targeting ORF3a, implicated
in
induction of cell apoptosis, both components playing a pivotal role in the pathogenicity
of this deadly coronavirus.
The majority of mutations focus on 6 viral proteins: S (21.8%
as of November 2020), N (18.1%), ORF3a (7.0%) and polyprotein
components Nsp12 (Pol), Nsp3 (PL-PRO) and Nsp2 (16.4%, 7.9%,
6.8% respectively). A closer inspection of the individual
viral component profiles often highlights mutation hotspots,
which could influence drug discovery decisions as presented
above. Here we focus on the Spike glycoprotein and the Nsp12
polyprotein component (Pol), which are currently targeted by
approved therapeutics and vaccines. Mutational profile of the
Spike glycoprotein are available
here.
Mutational profile of the Spike glycoprotein
As of November 2020, 261,401 mutations were targeting the
Spike glycoprotein. The vast majority map on 3D structure,
most targeting protein binding interfaces:
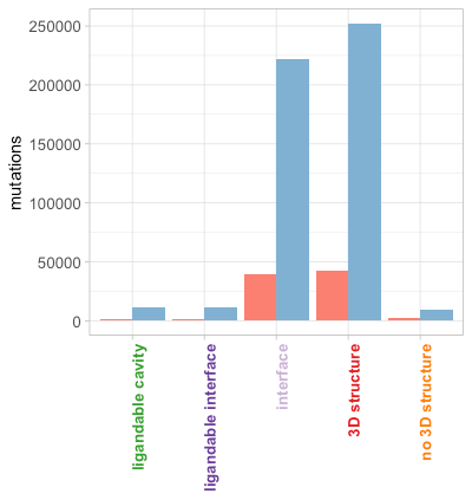 Spike glycoprotein mutation counts
Spike glycoprotein mutation counts
Significantly, mutations have started emerging in ligandable
cavities and interfaces, though their rates are currently
quite low. A closer inspection reveals that the dominant
mutation is indeed the D614G amino acid change which results
in increased ACE2 binding and fusion (Yurkovetskiy et al., 2020, Teruel et al -
preprint DOI,
Published). This mutation was already prevalent, yet not as a dominant
proponent in the June 2020 mutational landscape:
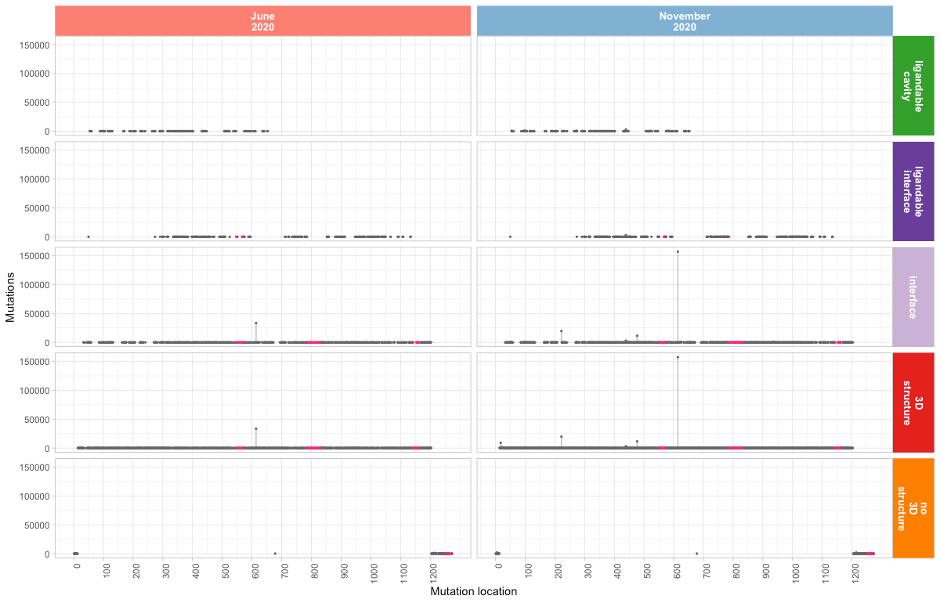 Spike glycoprotein mutation lollipop plot
Spike glycoprotein mutation lollipop plot
Residues 331 – 524 in the Receptor Binding Domain (RBD) are
not targeted as extensively by mutations. These residues form
the basis of the Pfizer mRNA vaccine. A number of other highly
immunogenic epitope sites, proposed by
Shrock et al., 2020, also exhibit far lower mutation rates and are highlighted
in magenta in the above lollipop plots.
Mutational profile of Nsp12 (Pol)
The Polymerase component of SARS2 harbours 196,086 mutations
as of mid-November 2020 (16.4% of all mutations). The most
dominant mutation, P323L, accounts for 80% of all Nsp12
mutations:
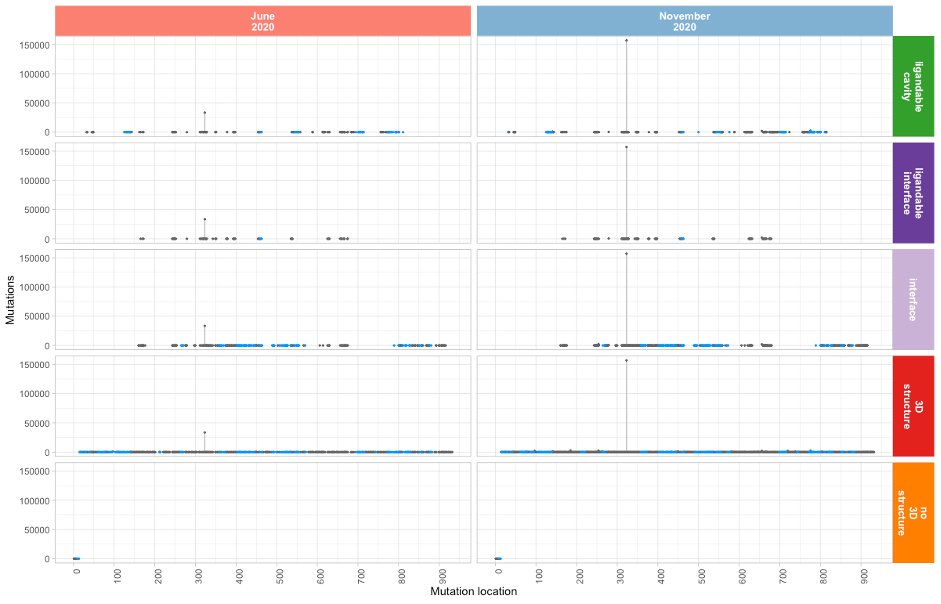 Nsp12 (Pol) mutation lollipop plot
Nsp12 (Pol) mutation lollipop plot
Proline 323 forms part of the Nsp8 binding interface and
participates in Hydrogen bonding with the Nsp8 Asparagine
residue 118. Note that a number of epitopes have been
identified by
Shrock et al., 2020
using triple Ala mutagenesis (shown in blue in the above
lollipop plot), presenting opportunities in the Nsp12 binding
interfaces with Nsp7 and Nsp8.
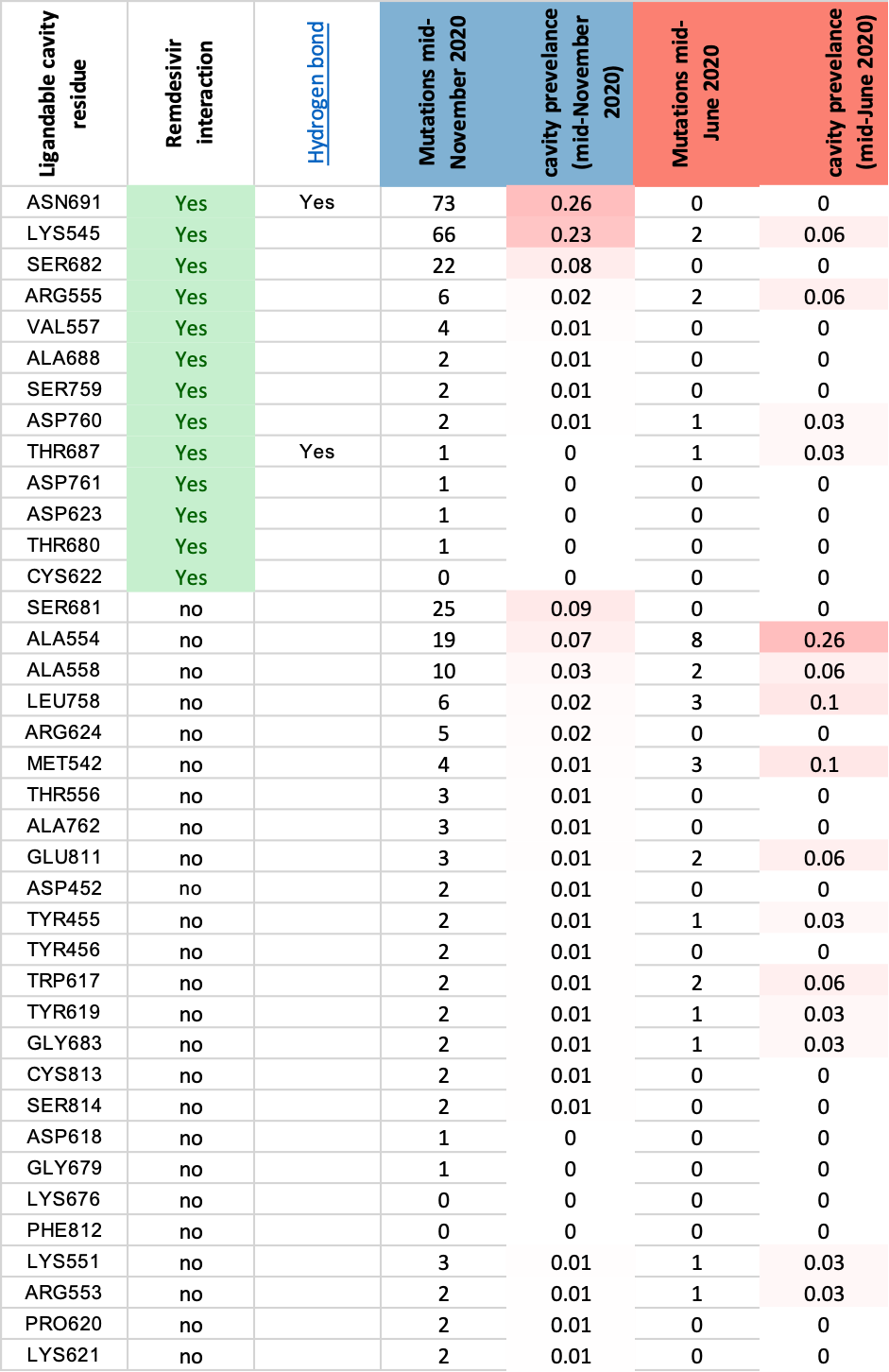
The Remdesivir ligandable cavity identified by canSAR-3D
comprises 38 amino acids exhibiting comparatively low mutation
rates (see table below). However there are mutations emerging
in residues N691, K545 and S682 which are important for
Remdesivir binding:
 Remdesivir cavity binding
Remdesivir cavity binding
 Mutation incidence within canSAR-3D identified
Remdesivir binding ligandable pocket
Mutation incidence within canSAR-3D identified
Remdesivir binding ligandable pocket
The canSAR-3D ligandable cavity residues are shown as a
molecular surface, using a rainbow spectrum for indicating
mutation rates (blue = low, red = high). The Remdesivir
molecule is shown in red, and the three most significantly
mutated residues are highlighted.
Following the recent FDA approval of Remdesivir for covid19
treatment it will be important to monitor whether the mutation
rate in this cavity increases.
The next full mutational profile update for SARS-CoV-2 is
scheduled for February 2021, to mark the first year of the
covid19 pandemic.